Research Programme: Genetics and Genomics hereditary cancer
Principal Investigator: Prof. Giuseppe Novelli

Most cancers occur “sporadically,” however, ~4-17% of cancers are inherited. Several hereditary cancer syndromes are autosomal recessive, influencing significantly risk assessment and genetic counseling in at risk families. Genetic testing is playing an increasing role in the evaluation and management of patients at increased risk of developing cancer. The knowledge of a pathogenic mutation prior to the onset of cancer can inform screening, result in or early cancer detection, and save lives. Moreover, cancer patients’ therapy can be tailored based on genetic test. e.g. TP53, BAP1, PTEN, able to influence therapy, since when these patients develop early stage breast cancer, mastectomy is preferred over lumpectomy followed by radiation therapy, because ionizing radiation may cause secondary malignancies in carriers of TP53 and BAP1 genetic mutations. Similarly, in the light of promising effects of PARPi therapy in BRCA positive ovarian and breast cancer patients, clinical trials on PARPi treatment in patients with mesothelioma could be warranted. In the long term, oncology patients’ survivorship plans can be tailored to incorporate surveillance and prevention for secondary malignancies, as many of these patients develop multiple cancers during their lifetime. Recent genetic progress provide the tools to assist in planning a life-long series of interventions including genetic counselling, prevention, surveillance and early detection that is often life saving and/or prolong life expectancy: for example, removal of an early stage melanoma in carriers of germline CDKN2A or BAP1 mutations, because of close monitoring of these high-risk individuals. An high frequency of pathogenic germiline variants has been detected in the homologous recombination pathway in patients with advanced solid cancers with potential PARP inhibitor sensitive tumors. Families with affected members carrying POLE or POLD1 variants show an high CRC risks. The importance of germline variants in the DNA repairome is not confined to the colorectal, but it has been shown in other tumors, such as breast/ ovarian and prostate. Repair DNA process is a fundamental property of all living organisms. DNA polymerase delta (Pol δ), encoded by POLD1 gene, plays an important role in genome maintenance through its involvement in synthetic repair processes. Polδ has a primary role as replicative polymerase for the lagging strand and an important proofreading ability conferred by the exonuclease activity. Reduced expression or activity of POLD1 correlates with the decrease of DNA repair capacity in senescent cells and normal aging. Damaging mutations in the proofreading domain of POLD1 as the underlying cause of early-onset colorectal, endometrial and breast inherited cancers, and suggested that mutations in POLD1 may influence therapeutic management. In addition, mutations in POLD1 have been identified in the developmental MDPL disorder characterized by premature aging features. Cells harboring the recurrent heterozygous in-frame p.Ser605del deletion showed nuclear alterations and apoptosis, strongly linked to genomic instability. More, DNA damage-induced treatment in MDPL fibroblasts revealed a poor capacity of DNA damage repair, the presence of micronuclei, cell cycle arrest. The further investigation of the functional linkages between POLD1, cancer, and aging may reveal shared and divergent pathways to improve clinical practice, drug treatments, and use of genome-editing technologies. We AIM to identify the:
a) proportion of clinically actionable mutations in patients/family to various cancer syndromes;
b) association of mutations with therapeutic management for prevention of family members;
c) prevalence of germline mutations in a sub-group of patients as pilot or large screens.
EXPERIMENTAL DESIGN. A total of 300 individuals will be tested, selected according to specific selected criteria (familial cancer, predisposing mutations, microsatellite instability, etc.), using a targeted tumor-normal sequencing panel, to identify carriers of germline mutations that predispose to cancer. Annotation, interpretation of all identified variants, bioinformatics, clinical significance of all identified variants and genetic counseling will be provided by expert geneticists to all subjects tested and their relatives.
CONCLUSION & PERSPECTIVE. We will identify clinically significant heritable mutations in selected population of patients with advanced cancer; development of selective therapeutic protocols; identification of pathogenic variants; get genomic variation vs diseases relationships.
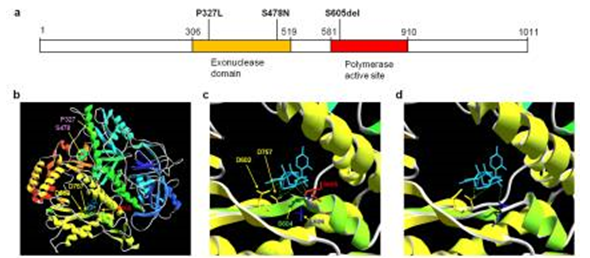
(a) POLD1Gene. Cancer mutations (P327L and S478N) affect residues within the exonuclease (proofreading) domain whereas the S605del is within the polymerase active site. (b) POLD1 structure. (c) wild type POLD1, and (d) mutant S605del. Note the predicted deformation of the catalytic domain and loss of hydrogen bonding to substrate in S605del compared to normal POLD1.