Research Programme: Cell Death and Autophagy Mechanisms
Principal Investigator: Prof. Mauro Piacentini

The scientific interests of Prof Piacentini focused on the dissection at the molecular level of two basic cellular events: cell death and autophagy. Initially, he has been investigating the molecular mechanisms involving the Type 2 transglutaminase (TG2) in the regulation in the skin terminal differentiation from which he moved to study the biological role of this enzyme in the process of apoptosis. For these studies Prof. Piacentini has received in 2007 the prestigious “Descartes” award from the European Commission. Recently, in the laboratory of Prof. Piacentini has been discovered one of the essential member of the Beclin1 complex regulating autophagy, Ambra1.
Type 2 Transglutaminase
Transglutaminase type 2 (TG2) is a multifunctional ubiquitously expressed member of TG family that catalyzes post-translational modifications of proteins through both Ca2+ dependent and independent reactions (3,4). Several unique features, including its ubiquitous expression, widespread localization, binding to and hydrolysis of guanine nucleotides, distinguish TG2 from the other transglutaminases. In fact, TG2 might also act as a GTP−binding protein that mediates intracellular signalling by coupling the alpha−1 beta−adrenergic receptor to the phospholipase C−gamma1. Substantial evidences indicate that under physiological circumstances the enzyme may also act as protein disulphide isomerase (PDI) (4). It has been established that TG2 switches its 3D structure from a nucleotide-bound “closed” to the transamidation/PDI prone “open” conformation In fact, in low intracellular nM Ca2+ concentrations, it mainly acts as a GTPase-supporting growth. By contrast, when cells are injured and the intracellular Ca2+ concentration increases, it acts as a crosslinking or PDI enzyme playing a key role in autophagy and if the cellular damage is irreversible inducing apoptosis. We have recently demonstrated that TG2 regulates cellular proteostasis and cell’s adaptation to stress by acting as a PDI (1, 2). At the molecular level our preliminary results indicate that the TG2’s PDI activity is responsible for the trimerization and activation of the heat shock factor 1 (HSF1), the main transcriptional factor regulating the inducible heat shock response. We have also recently demonstrated a key functional role of TG2 in the regulation of the mitochondrial/endoplamic reticulum contact sites. In fact, TG2 interactome analysis reveals an enzyme interaction with GRP75 (glucose-regulated protein 75). GRP75 localizes in mitochondria-associated membranes (MAMs) and acts as a bridging molecule between the two organelles by assembling the IP3R-GRP75-VDAC complex, which is involved in the transport of Ca2+ from the endoplasmic reticulum (ER) to mitochondria. The absence of the TG2-GRP75 interaction leads to an increase of the interaction between IP3R-3 and GRP75; a decrease of the number of ER-mitochondria contact sites; an impairment of the ER-mitochondrial Ca2+ flux; and an altered profile of the MAM proteome.
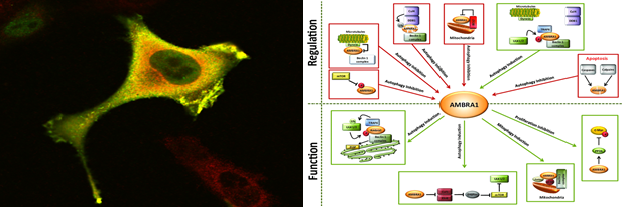
Ambra1 and autophagy regulation
Autophagy is a self-degradative process involved both in basal turnover of cellular components and in response to nutrient starvation or organelle damage in a wide range of eukaryotes (1). We showed that Ambra1 (activating molecule in Beclin1-regulated autophagy), a large, previously unknown protein bearing a WD40 domain at its amino terminus, regulates autophagy by interacting with Beclin1 (1). Ambra1 functional deficiency in mouse embryos leads to severe neural tube defects associated with autophagy impairment associated with accumulation of ubiquitinated proteins. Indeed, we reported that the E3 ubiquitin ligases Cullin-5 and Cullin-4 regulate the onset and termination of autophagy, respectively, by dynamically interacting with AMBRA1 (2). Under normal conditions, Cullin-4 binding to AMBRA1 and limits its protein abundance. Autophagy stimuli promote AMBRA1 stabilization by causing ULK1-dependent Cullin-4 release. Cullin-4/AMBRA1 dissociation is transient, and the re-established interaction triggers AMBRA1 degradation, terminating the autophagy response. Recently, we applied our knowledge about the role E3 ubiquitin ligases to unveil the role of autophagy in specific myopathies such as LGMD2H muscle dystrophy. We showed that TRIM32 is required for the induction of muscle autophagy in atrophic conditions and its inhibition results in a defective autophagy response to muscle atrophy. The pro-autophagic function of TRIM32 relies on its ability to bind the autophagy proteins AMBRA1 and ULK1 and stimulate ULK1 activity via unanchored K63-linked polyubiquitin (5).